Understanding CTD Module 2 and Its Role in FDA Submissions
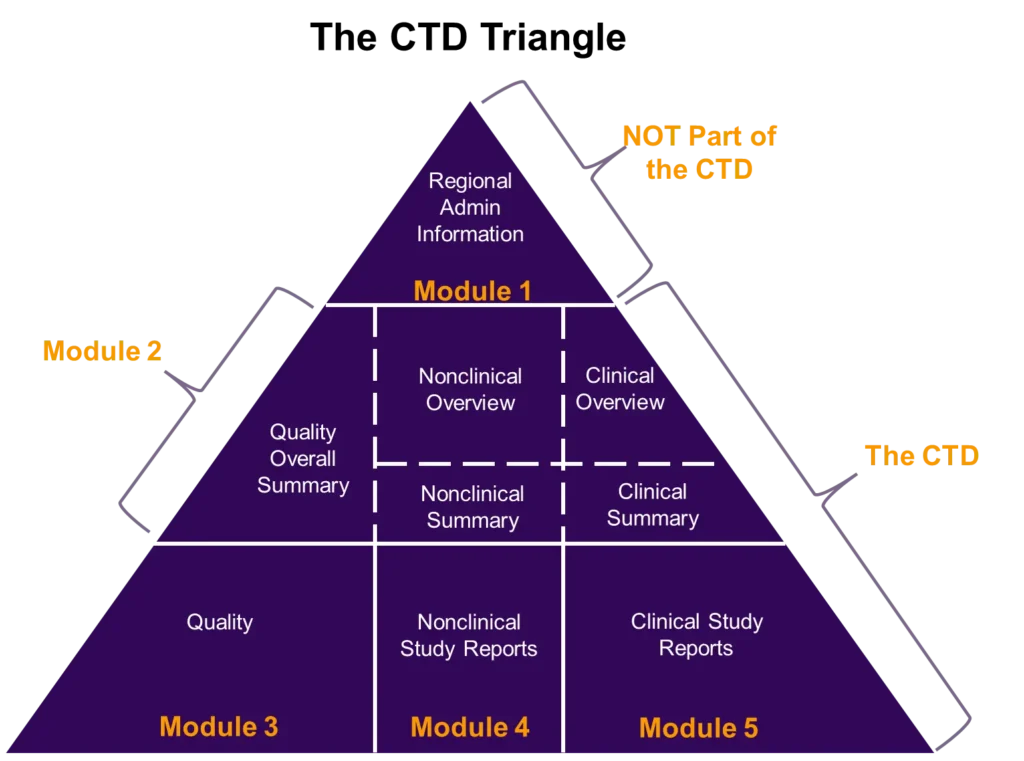
The Common Technical Document (CTD), written by a multidisciplinary team of scientific and regulatory experts, provides a universal format for regulatory submissions (i.e., INDs, NDAs, BLAs, and ANDAs) to the FDA. CTD Module 1 contains region-specific administrative information, CTD Module 2 contains manufacturing, nonclinical, and clinical overviews and summaries, and Modules 3 to 5 contain detailed manufacturing information, nonclinical, and clinical study reports (figure 1). For clinical pharmacologists, CTD Module 2 is where we describe the pharmacokinetics (PK) and pharmacodynamics (PD) of a drug. Two sections specifically, 2.7.1 (Summary of Biopharmaceutics Studies and Associated Analytical Methods) and 2.7.2 (Summary of Clinical Pharmacology Studies) require scientific expertise in clinical pharmacology and biopharmaceutics and often include information highly relevant for drug labeling.
To author Sections 2.7.1 and 2.7.2, it is essential that complex quantitative data is effectively translated into a story that regulatory agencies can easily follow.
Section 2.7.1: Summarizing Biopharmaceutics Studies and Analytical Methods
Biopharmaceutics studies bridge chemistry, manufacturing, and control (CMC) data with clinical performance. The 2.7.1 section should therefore provide a factual summary of all in vitro dissolution, bioavailability (BA), and bioequivalence (BE) studies carried out with the drug substance or drug product. Food effect studies, which are commonly conducted as part of Phase 1, are also frequently included along with BA/BE studies. Section 2.7.1 results focus on how the drug formulation performs, whether the intended dose delivers the expected exposure, and whether BA or BE has been demonstrated across studies and formulations, with particular attention to differences in results across studies. Examples of content that may be summarized in Section 2.7.1 include the following:
- Food effect study examining the impact of a high-fat meal on PK
- Relative bioavailability study comparing the PK of an older formulation versus a new (to-be-marketed) formulation
- All types of formulations and batches used in each clinical study
- In vitro data:
- Dissolution at different dosage strengths
- Solubility at different pH values
- Permeability across Caco-2 cells
- Bioanalytical methods for parent drug and metabolite for all clinical studies
- Bioanalytical methods for immunogenicity evaluation
The FDA, through its 2019 bioanalytical template guidance requested tabular summaries of bioanalytical information for all clinical studies in a program (not just biopharmaceutics studies). This includes tables containing bioanalytical method life cycle information, summary method performance, and summary of method modifications and cross-validation results. The completion of these tables is contingent on finalized bioanalytical reports for all clinical studies and requires an in-depth understanding of the bioanalytical data. Ultimately, the information on formulations and bioanalytical methods presented in 2.7.1 allow regulatory reviewers to trust the PK data summarized in the next section, 2.7.2.
Section 2.7.2: Translating Clinical Pharmacology Data into Regulatory Insights
Section 2.7.2 of CTD Module 2 provides an overall view of clinical pharmacology studies as well as any other studies that have collected PK or PD data. Additionally, in vitro studies performed with human cells, tissues, or biomaterials that are pertinent to PK processes are included. This section generally provides a tabular listing of all clinical pharmacology studies together with a narrative description of the relevant features and outcomes of each of the critical individual studies that provided in vitro or clinical data and information relevant to PK, PD, and PK/PD relationships. Frequently, PK information presented in this section includes dose proportionality, accumulation, and time to reach steady state. Examples of content that may be summarized in Section 2.7.2 include the following:
- Single ascending dose (SAD)/ multiple ascending dose (MAD) study (typically first-in-human) looking at a range of doses
- Mass balance and metabolite identification (also known as absorption, metabolism, distribution, excretion [ADME] or absorption, metabolism, excretion [AME] studies) in male healthy participants
- Drug-drug interaction (DDI) evaluation comparing the PK of a drug with or without coadministration of a strong cytochrome P450 (CYP)3A4 inhibitor
- Organ impairment study examining the impact of renal impairment on PK
- Exposure-response modeling and population PK (popPK) analyses integrating multiple different studies
- Special studies (e.g., microbiology or immunogenicity)
This section, along with 2.7.1 of CTD Module 2, translates quantitative science into clinical decision-making: dose to achieve safe and therapeutic exposure, need to / how to adjust dose for organ impairment, administration in regard to food, and what drug interactions (if any) warrant caution or contraindication. A large portion of the label is based on clinical pharmacology studies, and the information presented in 2.7.1 and 2.7.2 often directly corresponds to the label, and thus also to dosing patients in the clinical setting.
Key Challenges in Authoring Sections 2.7.1 and 2.7.2
Authoring Sections 2.7.1 and 2.7.2 is often a substantial effort, depending on the type and amount of content included. For example, Section 2.7.2 can be hundreds of pages including tables if there are a large number of studies and/or if there are special studies with microbiology and immunogenicity data included. A single 2.7.2 section may integrate dozens of studies (clinical and human biomaterials) and thousands of PK observations and model parameters. Additionally, the bioanalytical tables recommended to be included in Section 2.7.1 per FDA guidance can be very lengthy, especially in programs with numerous bioanalytical and validation reports. Compiling these tables requires specialized bioanalytical expertise to not only locate the right data to include in the right place within the tables, but also to verify that all numbers have been confirmed against source records and that no information is missing.
How Sections 2.7.1 and 2.7.2 Support Other CTD Components
The content of 2.7.1 and 2.7.2 helps provide a foundation for other sections to build upon, including the Integrated Summary of Safety (ISS), Integrated Summary of Efficacy (ISE), and Integrated Summary of Immunogenicity (ISI), which are generally included in module 5. For both ISS and ISE, drawing on pertinent clinical pharmacology data including PK, popPK, and PD results can be key in clarifying the exposure-response relationship of a drug. In the ISI, bioanalytical and PK information on immune response (i.e., anti-drug antibodies [ADAs]) aligns with the information summarized in 2.7.1 and 2.7.2, respectively. Because ADAs can alter the PK, PD, safety, and efficacy of a drug, describing these relationships in the ISI is necessary to fully characterize the impact of immune response on a drug.
Best Practices for Writing Clear, Reviewer-Friendly Summaries
Although accurately reporting scientific findings is paramount when summarizing biopharmaceutics and clinical pharmacology data in a regulatory submission, writing in a style to easily facilitate review is also important.
Suggested approaches for authoring quality CTD summary sections include the following:
- Starting with Key Takeaways – Summarize the most critical findings upfront.
- Using Cross-Referencing – Link to other sections within each document and to other modules as needed instead of repeating data.
- Choosing Relevant Tables and Figures – Avoid overwhelming reviewers with unnecessary visuals.
- Ensuring Consistency – Align with Module 2.5 (Clinical Overview) and other sections for seamless review.
- Writing for Review Efficiency – Use clear headings, concise language, and logical flow.
Why Expertise Matters
Sections 2.7.1 and 2.7.2 of CTD Module 2 form the backbone of many NDA submissions. Authoring these sections requires mastery of PK/PD principles and strategic insight to know how findings influence labeling and clinical use beyond just a summary of results. Behind every concise paragraph lies an immense amount of data, interpretation, and scientific expertise.
To learn more about CTD, regulatory submissions, technical documentation, and other ways we can support your drug development and clinical pharmacology studies, visit www.allucent.com or ask us your questions.
Relevant articles
eCTD Submission: FDA Guidelines & Common Mistakes
9 NDA Submission Tips for a Successful Application
References
ICH – M4E(R2): The CTD – Efficacy Guidance for Industry, 2017. https://www.fda.gov/media/93569/download
FDA – Bioanalytical Methods Templates Technical Specifications Document, 2019. https://www.fda.gov/media/131425/download
Request a Proposal
Share This