
In a recent blog post, I summarized a revised FDA guidance issued in January 2019, Rare Diseases: Common Issues in Drug Development.1 The guidance covers a range of topics, including the use of surrogate biomarkers, nonclinical flexibility, and natural history studies, to assist sponsors in conducting more efficient drug development programs for rare diseases. In March 2019, FDA released its sixth rare disease guidance, Rare Diseases: Natural History Studies for Drug Development,2 which expands on the topic of natural history studies and how they can be used to support the development of drugs and biologics. Due to the limited information on the natural histories of many rare diseases, natural history studies are particularly important for the development of rare disease therapeutics. Natural history studies provide valuable knowledge of a disease that can aid in the design of clinical trials. Further, they may also provide data to be used as an external control in a clinical trial when a placebo or active-control arm may not be feasible or ethical. Major highlights of the March 2019 guidance are summarized below. What is a natural history study? A natural history study is an observational study that follows the course of a disease in individuals to better understand how a disease develops and how it may be treated. Natural history studies aim to identify demographic, genetic, or environmental variables that correlate with the disease’s development and outcomes. Although the natural history of a disease is defined as the course a disease takes in the absence of intervention, natural history studies often include patients receiving the current standard of care, which may alter how the disease naturally progresses. How can natural history studies be used in drug development? FDA’s guidance highlights four key ways in which a natural history study may contribute to a drug development program:
- Identification of the patient population
- Identification or development of clinical outcome assessments
- Identification or development of biomarkers
- Design of externally controlled studies
While not the primary focus of the guidance, FDA touches briefly on the use of natural history data as an external control in clinical trials. The use of natural history data as an external control is of particular interest to sponsors developing therapies for rare diseases, for which placebo-controlled studies are difficult to complete due to ethical concerns or subject limitations, such as small populations. FDA recognizes that a natural history study may provide an external control group for interventional trials if it is well-designed and conducted. However, natural history studies are subject to certain biases (see Table 1), which can limit their ability to demonstrate effectiveness. To mitigate bias, FDA notes that the treatment effect of the investigational drug should be dramatic. FDA distinguishes between two types of external controls: nonconcurrent and concurrent (Figure 1). A natural history study completed before an interventional trial is classified as a nonconcurrent external control (also commonly referred to as a historical control). Figure 1. Types of External Controls 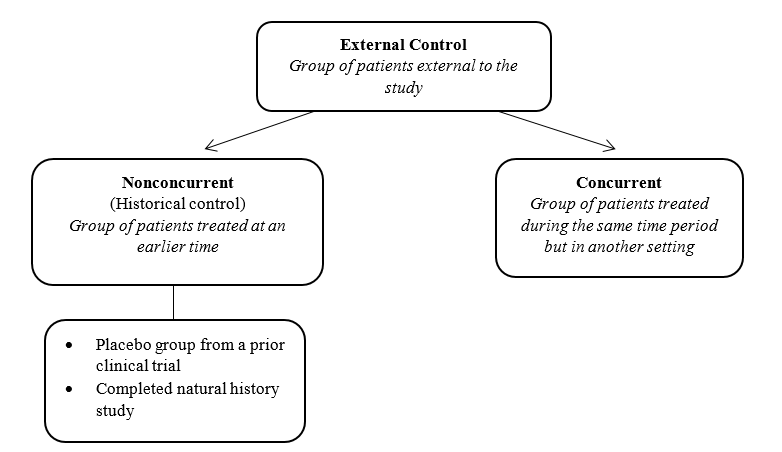 What are the different types of natural history studies? As noted above, natural history studies can play an important role in the development of rare disease therapies. Depending on the goal of the drug development program, the design of a natural history study can take on several forms. Natural history studies can be:
What are the different types of natural history studies? As noted above, natural history studies can play an important role in the development of rare disease therapies. Depending on the goal of the drug development program, the design of a natural history study can take on several forms. Natural history studies can be:
- Retrospective or prospective; and
- Cross-sectional or longitudinal
Retrospective studies are studies in which patient evaluations have already occurred. In contrast, prospective studies are studies in which evaluations occur in the future according to a prespecified data collection plan. In cross-sectional studies, data are collected from patients at a single point in time, which may be set by a stage of illness, date of diagnosis, onset of symptoms, or other criteria. In longitudinal studies, data are collected from the same group of patients over a period of time. Table 1 compares the characteristics of retrospective studies with those of prospective studies. Table 1. Comparison of Retrospective and Prospective Studies
| Topic | Retrospective | Prospective |
| Data collection | Data commonly come from existing medical records (e.g., patient charts) Data may have been collected at variable time points or were obtained inconsistently | New data are generated after initiation of the study Studies can follow standard operating procedures, which allows for greater consistency in the information collected |
| Duration | Studies can be performed quickly | Studies generally require more time |
| Medical terminology | Medical terminology may have changed over time or have been used inconsistently among health care providers (unclear use of terms may limit interpretability and result in incomplete information) | Studies can use up-to-date definitions of medical conditions and treatments |
| Bias | Can be biased through patient selection criteria and through selection of dates of inception and cutoff May be subjected to referral bias (study includes only the most severely affected patients) May be subjected to length-biased sampling (patients who have been in the database the longest may be overrepresented) | Study evaluations occur in the future and are not highly susceptible to bias |
Due to the limitations of retrospective studies, which may include inconsistent measurement procedures, irregular time intervals, and the unclear use of terms, retrospective natural history studies are more susceptible to bias than prospective natural history studies. These limitations may prevent such studies from being used as external controls if patient characteristics of an interventional trial cannot be matched with those of the historical control. In contrast, prospective studies allow for the use of standard operating procedures and schedules so that data is collected consistently. Retrospective and prospective studies may be further classified as either cross-sectional or longitudinal. Table 2 summarizes the advantages of cross-sectional and longitudinal studies. Table 2. Comparison of Cross-Sectional and Longitudinal Studies
| Advantage | Cross-Sectional | Longitudinal |
| Faster data collection and analysis? | ✔ | |
| Less resource intensive? | ✔ | |
| Provide more comprehensive information about disease onset and progression? | ✔ | |
| Better suited to distinguish the variety of phenotypes and subgroups of a disease? | ✔ | |
| Data are more likely to be used an external control group? | ✔ |
While data from cross-sectional studies are less likely to be suited to be used as an external control group compared to longitudinal studies, cross-sectional natural history studies may provide valuable information for therapies intended to provide largely immediate benefits in patients experiencing an acute episode or flare of a disease. In general, longitudinal studies provide more comprehensive information about disease onset and progression. It is important to note that while prospective, longitudinal natural history studies are desirable, the initiation of such studies should not delay a drug development program if interventional testing is ready to proceed for a serious disease with unmet medical need. What are important considerations for the protocol, study design, data collection, and protection of human subjects in natural history studies? FDA’s Natural History Studies guidance2 also includes sections on topics related to the protocol, study design, data collection, human subject protection, and interactions with the FDA. Below is a subset of the considerations for natural history studies noted in the guidance:
- Data that will be used to support a marketing application should be collected according to the data standards for marketing applications. International data standards should also be considered.
- Data should not be limited to the most severely affected body systems because treatment responses might be more reliably detected by evaluation of a less affected body system.
- Natural history studies should have a prospectively defined statistical analysis plan.
- Patients may be identified via disease-specific support groups or patient advocacy groups.
- Natural history studies can be registered in https://www.ClinicalTrials.gov to increase participation and recruitment.
- Natural history studies should code data from patient experiences using a vocabulary that is standardized (e.g., Medical Dictionary for Regulatory Activities [MedDRA]).
- FDA encourages the dissemination of clinical data from a natural history study, as well as the methods and practical aspects of conducting the study, as widely as possible (e.g., through peer-reviewed publications) due to the lack of information available for rare disease.
- Natural history studies may be subject to 21 CFR Parts 50 and 56 if they meet the definitions of clinical investigation and other applicable definitions under those parts. Regulations under 45 CFR Part 46 (also known as the Common Rule) may also apply.
- Institutional Review Board (IRB) review is generally required for a natural history study that is subject to the Common Rule.
- When planning a natural history study, it is important to consider that the data and biospecimens collected may be useful in future secondary research. The study organizer should work with an IRB to determine the best approach to obtain consent for this possibility.
- FDA discussions do not need to be conducted in the context of a regulatory submission or meeting. Critical Path Innovation meetings may provide advice on drug development issues.
References 1FDA. Rare Diseases: Common Issues in Drug Development. Available at: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM629579.pdf; 31 January 2019. 2FDA. Rare Diseases: Natural History Studies for Drug Development. Available at: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM634062.pdf; 22 March 2019.
Request a Proposal
Share This