Brian Abbott, MD
Executive Medical Director,
Therapeutic Area Medical Lead,
Oncology/Hematology
Oncology remains one of the most challenging therapeutic areas, with cancer clinical trials facing the lowest success rates. This underscores the need for innovative strategies and an oncology CRO partner who understands the complexities of this field and has deep experience across all stages of oncology drug development.
At Allucent, we are more than just an oncology CRO, we are your strategic partner in navigating the intricate landscape of oncology drug development. With expertise across all development phases from preclinical to post-marketing, we provide tailored solutions to overcome regulatory hurdles, patient recruitment challenges, and evolving research demands.
From early to late-stage cancer clinical trials, we drive efficiency, compliance, and data-driven success helping you bring life-changing therapies to patients faster.

Oncology clinical trials often face recruitment challenges due to stringent eligibility criteria and the need for specific patient populations. At Allucent, we address these issues by leveraging global reach, genetic data, and electronic medical records to identify the right patients quickly and efficiently.
As a specialized oncology CRO, we offer tailored solutions for oncology clinical research, supporting both hybrid and on-site oncology trials. Our expertise ensures streamlined recruitment while actively engaging patient communities to boost participation and retention.
Learn More

Oncology clinical trials require specialized expertise and complex study designs. As a trusted Oncology CRO, we excel in oncology research, implementing adaptive designs, basket trials, crossover designs, and single-arm trials to ensure precision and flexibility.
Our biostatistics and regulatory teams provide strategic guidance on cancer clinical trials, optimizing endpoint selection, data strategy, trial design and regulatory compliance. We also incorporate biomarkers into study designs to enhance precision medicine approaches, allowing for more targeted and effective treatments.
Learn More
At Allucent, we help you navigate the high costs and site saturation challenges by leveraging our extensive network of sites and strong relationships with key opinion leaders (KOLs). Our deep expertise in oncology clinical research and strong relationships allow us to gain valuable insights into site capabilities and patient recruitment potential, ensuring your cancer clinical trial is conducted at the most suitable locations.
Our expertise in conducting thorough feasibility assessments helps identify the right sites, avoiding oversaturated locations and optimizing trial execution. With our optimized site management and ongoing support, we ensure high-quality results while managing costs effectively.
Learn More

Navigating the regulatory complexities of oncology clinical research and drug development requires expert guidance. We provide strategic support from early planning to post-approval, ensuring your Oncology clinical trial meet regulatory standards while optimizing fast-track approvals.
Our expert team, including former regulators from the FDA, EMA, and other global health authorities, provides strategic support in study design, endpoint selection, and data strategy to help you overcome the unique challenges of oncology drug development. With expertise in Project Optimus and Project Orbis, we streamline cancer clinical trials, securing breakthrough designations and ensuring compliance.
Learn More

The Allucent Center of Expertise (ACE) for oncology and hematology is powered by a dedicated team of experts in this therapeutic area, offering specialized knowledge and cutting-edge solutions across the entire oncology drug development process. The oncology ACE combines our oncology and hematology clinical trial operations excellence with deep expertise in regulatory strategy, clinical pharmacology, and biostatistics, providing comprehensive support from preclinical through to clinical development and beyond.
Pioneering Cancer Therapies for Tomorrow’s Cures
We have extensive experience with both traditional and cutting-edge therapies, including molecularly targeted agents, immunotherapies, and cell and gene therapies. We also specialize in combination therapies, and have substantial experience with anticancer vaccines, antibodies (monoclonal, bispecific, and ADCs), nanoparticle formulations, and modified chemotherapies to improve efficacy and patient outcomes.
Click icons for more information:
Involvement in over 40 trials with autologous and allogenic cell products, including NK cells, T-cells, and oncolytic viruses. Multidisciplinary expertise in CGT trials.
Cell and Gene Therapy(CGT) trials are logistically challenging, requiring highly adept, multidisciplinary expertise. Allucent understands the unique needs of the small and mid-sized biopharma companies that are often at the forefront of development in this exciting field. Our project teams have been involved in more than 40 CGT studies and projects with both autologous and allogenic cell products. Besides trials that investigate natural killer (NK) cells, stem cells, dendritic cell, and T-cells, we have also worked on trials with oncolytic viruses. Learn more about out CGT expertise.
Supported regulatory approvals for radiolabeled proteins and Radium-223 in trials across Phases I-IV, working on NDA and MAA submissions.
We are highly experienced in performing trials with radiolabeled proteins and Radium-223 in neuroendocrine tumors and prostate cancer. Our team of experts works in the United States and Europe across Phase I-IV trials and has supported the regulatory approval of new drug applications (NDAs) at the FDA and marketing authorization applications (MAAs) at the EMA of radiolabeled proteins.
Extensive work with therapies targeting hormone-driven cancers like breast and prostate. Expertise in aromatase inhibitors, selective estrogen receptor inhibitors, and more.
Altering the levels or activity of certain hormones can cause some cancers to cease growth and/or undergo cell death. We have performed studies with aromatase inhibitors, selective estrogen receptor inhibitors, and synthetic gonadotropin releasing hormone agonists as single therapies but also as part of combination therapies.
Experience across Phases I-IV in combination therapies with targeted and immunotherapies, optimizing doses to reduce toxicity and improve outcomes. Over 20 studies in the past five years.
Advances in chemotherapy have evolved; it is no longer used solely as a monotherapy but can also be part of a combination therapy. Our clinical team has experience in chemotherapy combinations with targeted therapies and immunotherapies. In the last five years, our experts worked on over 20 studies ranging from Phases I to IV involving chemotherapy.
Experience with complex adaptive trial designs and basket trials, across more than 115 studies. Expertise with antibodies, small molecules, and FDA approvals of targeted therapies.
Our clinical team has worked with targeted therapies in Phase I to IV studies, encompassing complex adaptive trial designs and basket trials. We are experienced in mechanisms of action including the inhibition of angiogenesis, histone deacetylase (HDAC), integrin receptors, poly ADP ribose polymerase (PARP), growth factors, and the cell cycle, to name a few. Allucent has supported more than 115 studies and projects and worked with antibodies and small molecules such as protein kinase inhibitors.
Participation in over 100 studies, combining immuno-oncology (IO) drugs with chemotherapy and targeted therapies. Proficient in irRC, iRECIST, and immune-related adverse events.
Our experts have worked on more than 100 studies and projects in hematologic malignancies and solid tumors and are well-versed in the different immune-related tumor evaluation criteria such as irRC and iRECIST, as well as immune-related adverse events (irAE). We have in-depth knowledge and hands-on experience with Immuno-Oncology drugs including antibodies, peptides, oligonucleotides, cell-based products, oncolytic viruses, and vaccines.
Comprehensive Expertise Across Traditional and Advanced Therapies in Oncology

Combining clinical expertise with research is essential for driving innovation and effective therapies. By aligning with scientific advancements and regulatory standards, we streamline development, accelerating safe treatments in hematology and oncology. This integration ensures faster patient access to transformative care while maintaining the highest safety standards.
Oksana Fabri, PhD, MD
Sr. Medical Director, Therapeutic Area Medical Lead, Hematology/Oncology


We are specifically designed to provide hematology and oncology CRO services to small and mid-sized biotechs through every phase of drug development, from preclinical planning and IND submissions to the design and execution of Phase I-III trials, through marketing applications, to Phase IV and post-marketing initiatives to ensure ongoing success.
We have the hands-on knowledge to manage the complexities of cancer clinical trials and provide full-service support across various therapeutic areas. Our extensive experience across Early Phase through Phase III trials and beyond spans a wide range of oncology and hematology indications.
270+
Studies
4,000+
Sites
24,000+
Patients
40+
Countries
All-time years of experience
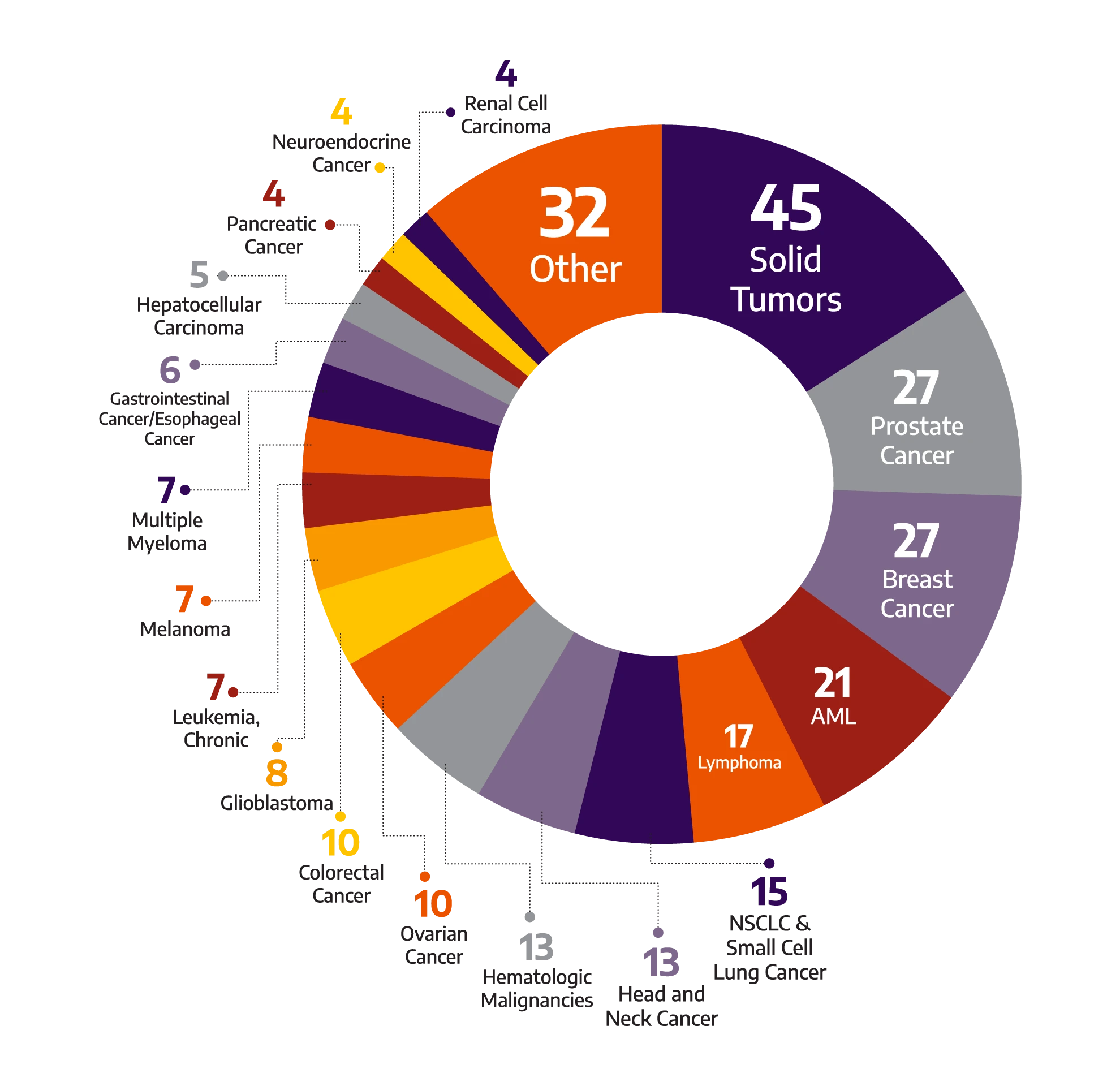
With global breadth and depth across a wide range of oncology and hematology indications, Allucent partners with biotech companies to bring innovative therapies to light.

Executive Medical Director,
Therapeutic Area Medical Lead,
Oncology/Hematology

Executive Medical Director,
Medical Affairs

Sr. Medical Director,
Therapeutic Area Medical Lead,
Hematology/Oncology

Global Operations Head,
Oncology/Hematology


Case Study
Learn More
Industry Featured Article
Learn More

White Paper
Learn More
Let us know how we can help you bring new therapies to light. Get in touch to get started.
Want to help small and mid-sized biotech companies change the therapeutic landscape?